

|
eparan sulfate structure and function 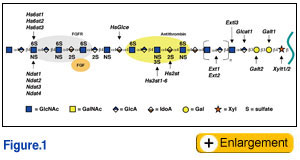 Heparan sulfate proteoglycans (HSPGs) are composed of a core protein and one or more heparan sulfate (HS) glycosaminoglycan (GAG) chains, a linear polysaccharide composed of alternating N-acetylated or N-sulfated glucosamine units (GlcNAc/GlcNS) and uronic acids (GlcA or IdoA). Three subfamilies of HSPGs exist: the membrane spanning proteoglycans (syndecans, betaglycan, and CD44v3), the GPI-linked proteoglycans (glypicans), and the secreted extracellular matrix proteoglycans (agrin, collagen XVIII, and perlecan). Hematopoietic cells also contain a secretory vesicle proteoglycan called serglycin. Some HSPGs contain chondroitin sulfate/dermatan sulfate, a related GAG chain that differs from HS in the component sugars (GalNAc and GlcA/IdoA) and patterns of modification, and other types of glycans (N-linked and O-linked mucin type chains).
Heparan sulfate proteoglycans (HSPGs) are composed of a core protein and one or more heparan sulfate (HS) glycosaminoglycan (GAG) chains, a linear polysaccharide composed of alternating N-acetylated or N-sulfated glucosamine units (GlcNAc/GlcNS) and uronic acids (GlcA or IdoA). Three subfamilies of HSPGs exist: the membrane spanning proteoglycans (syndecans, betaglycan, and CD44v3), the GPI-linked proteoglycans (glypicans), and the secreted extracellular matrix proteoglycans (agrin, collagen XVIII, and perlecan). Hematopoietic cells also contain a secretory vesicle proteoglycan called serglycin. Some HSPGs contain chondroitin sulfate/dermatan sulfate, a related GAG chain that differs from HS in the component sugars (GalNAc and GlcA/IdoA) and patterns of modification, and other types of glycans (N-linked and O-linked mucin type chains).
Because of their high negative charge, the HS chains bind to a large number of proteins, including most of the members of the FGF family and their receptor tyrosine kinases, TGFs, BMPs, Wnt proteins, chemokines, interleukins, as well as enzymes and enzyme inhibitors, lipases and apolipoproteins, and ECM and plasma proteins. The best studied example is the activation of the serpin protease inhibitor antithrombin by a specific pentasaccharide within heparin. Binding results in a conformational change and in the case of Factor IIa, approximation of the substrate with the inhibitor greatly accelerates antithrombin's ability to inactivate the protease. The interaction of heparin and HS with FGFs and their receptors has been characterized in great detail as well. In this case the GAG chain acts as a template that bridges FGF and FGFR (Fig. 1), like the interaction of heparin with antithrombin and Factor IIa. Formation of the complex effectively lowers the concentration of FGF required to initiate signaling through its receptor and extends the duration of the response.  Heparan sulfate and lipid metabolism Heparan sulfate and lipid metabolismIn the circulatory system, HSPGs modulate lipid metabolism by serving as receptors for tethering lipases to endothelial cell surfaces and as clearance receptors for lipoproteins in the liver. Lipoproteins transport cholesterol and triglycerides through the circulation. The triglyceride rich lipoproteins (TRLs) consist of chylomicrons, which arise from dietary lipids, and very low density lipoproteins (VLDL), which come from de novo synthesized lipids in the liver. Lipoprotein lipase (LPL) acts on these particles to generate free fatty acids for energy production in the heart and skeletal muscles and for storage in adipose tissue. The remnant particles are acted on further by hepatic lipase (HL) in the liver sinusoids, and are subsequently cleared by receptors in the liver, which recognize the apolipoproteins, apoB and apoE, or bound lipases. HS binds to several proteins important to lipoprotein metabolism, including apoB, apoE, LPL, and HL.  To study the role of hepatic and endothelial HS in lipoprotein metabolism, we altered the expression of the biosynthetic gene GlcNAc N-deacetylase/N-sulfotransferase (Ndst1) in a tissue specific manner using the Cre-loxP system, which resulted in ~50% reduction of sulfation of liver HS. Mice were viable and healthy, but they accumulated TRLs resembling chylomicron remnants and VLDL. Mutant mice synthesized VLDL normally, but showed reduced plasma clearance of VLDL and a corresponding reduction in hepatic VLDL uptake. Post-prandial studies showed that clearance of intestinally derived lipoproteins also was altered in the mutant. Compounding Ndst1-deficiency with a mutation in the LDL receptor enhanced the accumulation of TRLs and resulted in accumulation of cholesterol-rich particles as well, suggesting that HS participates in the clearance of cholesterol-rich lipoproteins as well. In contrast, when Ndst1 was inactivated in endothelial cells no change in plasma triglycerides or cholesterol occurred, suggesting that only hepatic proteoglycans were relevant in clearance. These findings show that under normal physiological conditions hepatic HSPGs work in parallel with LDL receptors to clear both intestinally-derived and hepatic lipoprotein particles.
These findings identify HSPGs as the long sought receptor for TRL clearance. To determine the relevant proteoglycans mediating plasma lipid clearance, RT-PCR analysis was performed on isolated hepatocytes. These cells express syndecans-1, -3, and -4, glypicans-1 and -4, and the matrix proteoglycans agrin, collagen XVIII, and perlecan. The HS chains on one or more of these proteoglycans bind to remnant particles presumably through associated apolipoproteins or lipases. Current studies focus on the identification of the relevant ligands and proteoglycan receptors, the mechanism of uptake of the particles, and structural determinants in the HS chains that mediate lipoprotein binding. To study the role of hepatic and endothelial HS in lipoprotein metabolism, we altered the expression of the biosynthetic gene GlcNAc N-deacetylase/N-sulfotransferase (Ndst1) in a tissue specific manner using the Cre-loxP system, which resulted in ~50% reduction of sulfation of liver HS. Mice were viable and healthy, but they accumulated TRLs resembling chylomicron remnants and VLDL. Mutant mice synthesized VLDL normally, but showed reduced plasma clearance of VLDL and a corresponding reduction in hepatic VLDL uptake. Post-prandial studies showed that clearance of intestinally derived lipoproteins also was altered in the mutant. Compounding Ndst1-deficiency with a mutation in the LDL receptor enhanced the accumulation of TRLs and resulted in accumulation of cholesterol-rich particles as well, suggesting that HS participates in the clearance of cholesterol-rich lipoproteins as well. In contrast, when Ndst1 was inactivated in endothelial cells no change in plasma triglycerides or cholesterol occurred, suggesting that only hepatic proteoglycans were relevant in clearance. These findings show that under normal physiological conditions hepatic HSPGs work in parallel with LDL receptors to clear both intestinally-derived and hepatic lipoprotein particles.
These findings identify HSPGs as the long sought receptor for TRL clearance. To determine the relevant proteoglycans mediating plasma lipid clearance, RT-PCR analysis was performed on isolated hepatocytes. These cells express syndecans-1, -3, and -4, glypicans-1 and -4, and the matrix proteoglycans agrin, collagen XVIII, and perlecan. The HS chains on one or more of these proteoglycans bind to remnant particles presumably through associated apolipoproteins or lipases. Current studies focus on the identification of the relevant ligands and proteoglycan receptors, the mechanism of uptake of the particles, and structural determinants in the HS chains that mediate lipoprotein binding.
Insulin-deficiency, hypertriglyceridemia and heparan sulfate Hypertriglyceridemia is a major complication of insulin-deficient diabetes that can lead to cardiovascular disease, the major cause of mortality in this patient population. Insulin-deficiency alters liver HS structure in both streptozotocin-induced and spontaneous rodent models of diabetes. In these models, the activity of Ndst1 appears to be reduced, but it remains unclear whether this change is responsible for the observed alteration of HS structure. Although it is tempting to speculate that a correlation exists, there are many enzymes involved in HS biosynthesis (Fig. 1) and the chains assemble on multiple core proteins (16 known). Based on these observations, we hypothesize triglyceride accumulation in diabetes results from altered binding of TRLs to liver HS (Fig. 3). One objective of our work is to determine the relevant biosynthetic enzymes and proteoglycan core proteins that are altered by insulin deficiency in a mouse model of Type I diabetes. Another goal is to determine the effects of reduced insulin and elevated glucose levels on HS biosynthesis in cultured hepatocytes. We also will attempt to restore HS synthesis in the liver of diabetic animals to determine if plasma triglycerides can be decreased. |
|||||||||||
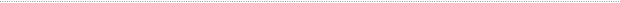 |
|||||||||||
| Reference: | |||||||||||
|
